
Main Market Comparability Of Key COVID-19 Laws – Anti-trust/Competitors Legislation
MAJOR MARKET COMPARISON OF KEY COVID-19 LEGISLATION
As pharmaceutical companies worldwide race to supply vaccines
and therapeutics to fight the spread of COVID-19, understanding the
laws and regulations that could impact parties involved in the
COVID-19 pandemic supply chain is increasingly important. This
article provides an overview and comparison of legislation relevant
to manufacturers, suppliers, distributors, and health professionals
involved in the response to the COVID-19 pandemic in each of the
United States (“US”), European Union (“EU”),
United Kingdom (“UK”), and People’s Republic of China
(“PRC”), including measures to ensure (1) immunity from
COVID-19 countermeasure liability; (2) government ability to direct
(or redirect) resources; (3) emergency use authorizations; (4)
price-gouging prevention; (5) cooperation between companies; and
(6) export controls.
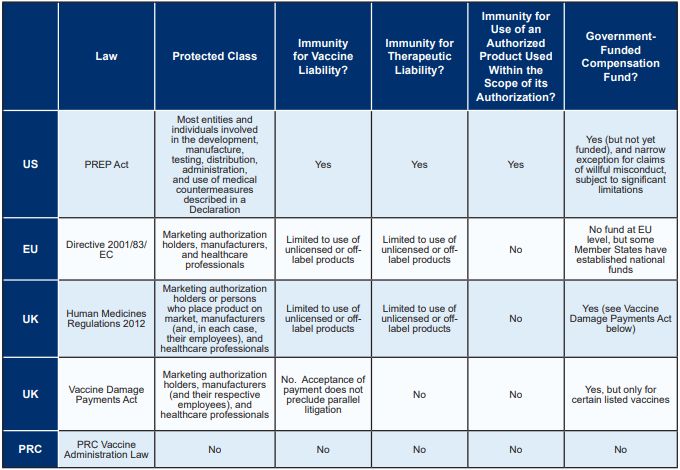
IMMUNITY FROM COVID-19 VACCINE AND THERAPEUTIC LIABILITY
The United States has the most comprehensive pandemic product
liability protection legislation of the major markets, affording
broad immunity to actors involved in the COVID-19 supply chain for
both vaccines and therapeutics. While the EU and UK provide limited
immunity for the use of unlicensed or off-label medicinal products,
the PRC does not provide any immunity for vaccine or therapeutic
liability. The following table compares immunity from
COVID-19-related liability in each of the major markets, as further
described below.
United States
The US Public Readiness and Emergency Preparedness
Act (“PREP Act”)
Enacted in 2005, the PREP Act authorizes the Secretary of the
Department of Health and Human Services (“HHS”) to
declare a public health emergency identifying “covered
countermeasures” and providing immunity against claims of loss
arising from such covered countermeasures. A PREP Act declaration
was issued on March 17, 2020, retroactive to the initial emergency
declaration on February 4, 2020, for activities related to
“Covered Countermeasures” against COVID-19, including
COVID-19 vaccines, therapeutics, medical devices, and diagnostics,
which has since been amended several times.1 In general,
such “Covered Countermeasures” are approved, cleared, or
licensed by the US Food and Drug administration (“FDA”);
authorized under an Emergency Use Authorization by the FDA;
authorized for investigational use, i.e., under an Investigational
New Drug (“IND”) or Investigational Device Exemption
(“IDE”); or otherwise permitted to be held or used for
emergency use in accordance with US Federal law.
The PREP Act, when invoked in a declaration under a pending
emergency, provides immunity for the manufacture, testing,
development, distribution, administration, and use of such covered
countermeasures against chemical, biological, radiological, and
nuclear agents of terrorism, epidemics, and pandemics-including
COVID-19. Individuals who suffer injuries from the administration
or use of products covered by the PREP Act’s immunity
provisions may seek redress from the Countermeasures Injury
Compensation Program (“CICP”), which is administered by
the Health Resources and Services Administration (part of the HHS).
The CICP is a “payer of last resort,” and any benefits
from the fund are reduced by the amounts payable by all other
public and private third-party payers (such as health insurance and
workers’ compensation).
Immunity protections are broad, and contrary state and local
laws and rulings are widely preempted; practically, the only time a
manufacturer of a COVID-19 covered countermeasure would not benefit
from PREP Act immunity would be if a suit is brought in the US
District Court for the District of Columbia by a plaintiff who has
suffered a serious injury or death, has rejected a payment from the
fund (which is not currently funded for COVID-19-related claims),
and has demonstrated by clear and convincing evidence that the
manufacturer engaged in “willful misconduct,” as defined
in the statute. It is worth noting that enforcement actions against
manufacturers of covered countermeasures regulated under the Public
Health Service Act or the Federal Food, Drug, and Cosmetic Act are
not considered willful misconduct unless the government initiates
an enforcement action that actually results in a criminal, civil,
or administrative penalty.
Unsurprisingly, the PREP Act does not provide immunity for
foreign claims where the US lacks jurisdiction. However, immunity
may be available for administration or use of a covered
countermeasure outside the US if the claim is based on events that
take place in the US or another link to the US makes it reasonable
to apply US law.
European Union
Directive 2001/83/EC of the European Parliament and
of the Council of 6 November 2001 on the Community code relating to
medicinal products for human use, as amended (“Directive
2001/83/EC”)
Directive 2001/83/EC was enacted in 2001 to consolidate earlier
EU legislation relating to medicinal products for human use and
remains the primary legislation governing the regulation of
pharmaceuticals in the EU. Directive 2001/83/EC and other EU
directives are transposed into the national laws of the EU Member
States and are implemented, applied, interpreted, and enforced by
their national competent authorities and courts as well as by the
European Court of Justice.
Like the PREP Act, Directive 2001/83/EC broadly applies to all
“medicinal products.” However, the scope of the immunity
afforded to persons involved in the COVID-19 supply chain is more
narrow than the PREP Act. Article 5(3) of Directive 2001/83/EC
requires Member States to put in place provisions to protect
marketing authorization holders, manufacturers, and healthcare
professionals from civil or administrative liability for any
consequences from the use of (1) an unauthorized medicinal product
or (2) the off-label use of an authorized medicinal product, if
such use was required or recommended by a competent authority in an
EU Member State in response to the COVID-19 pandemic. The scope of
such protection may vary between Member States depending on the
wording of national laws implementing Article 5(3) of Directive
2001/83/EC. There is no pan-EU compensation fund available with
respect to injuries experienced following receipt of a vaccine or
COVID-19; however, some individual EU Member States have
established funds that are available to their citizens at the
national level.
Furthermore, Article 5(4) of Directive 2001/83/EC states that
the limitation of liability provided under Article 5(3) does not
affect the manufacturer’s liability for defective products set
out in Directive 85/374/EEC (also known as the “Product
Liability Directive”). A medicinal product is considered to be
defective if it does not “provide the safety which a
person is entitled to expect, taking all circumstances into
account.” This is likely to apply, for example, in cases
of quality issues in the manufacturing of the medicinal product.
Liability under the Product Liability Directive is subject to a
number of defenses, including the so-called “development risks
defense,” which provides that a producer is not liable if it
can establish “that the state of scientific and technical
knowledge at the time when he put the product into circulation was
not such as to enable the existence of the defect to be
discovered.”
While governments in the EU have been resistant to any general
immunity for producers of medicinal products, including those
developed in a pandemic, the application of the Product Liability
Directive is likely to mitigate at least to some extent the risk of
a finding of liability. Therefore, the circumstances to be taken
into account when assessing a “defect” include whether a
product had been developed under accelerated timelines in a
pandemic situation (including potentially the fact that a marketing
authorization was granted on a conditional basis), in which case
the development risks defense is likely to apply. However, the
defenses available under the Product Liability Directive do not
protect producers against the resource and cost implications of
litigation.
United Kingdom
The Human Medicines Regulations 2012,2 as
amended (“Human Medicines Regulations” or the
“Regulations”)
The Human Medicines Regulations, established in 2012,
consolidated UK legislation relating to medicinal products for
human use in certain areas, including manufacturing, wholesale
dealing, and marketing authorizations. Regulation 345 of the
Regulations (which implements Article 5(3) of Directive 2001/83/EC
in the UK) protects marketing authorization holders or those
responsible for placing the product on the market, manufacturers
(and their respective employees), and healthcare professionals from
civil liability for loss and damage resulting from the use of an
unauthorized or off-label medicinal product if such use was
required or recommended by the UK licensing authority in response
to the suspected or confirmed spread of pathogenic agents, toxins,
chemical agents, or nuclear radiation that may cause harm to human
beings-including COVID-19.
The Human Medicines Regulations were most recently amended on
October 16, 2020 in response to the COVID-19 pandemic to, among
other things, expand the immunity from civil liability afforded to
healthcare workers and manufacturers to also include companies
producing the vaccine. Unlike the PREP Act, the Human Medicines
Regulations do not provide immunity for use of an authorized
product used within the scope of its authorization.
Vaccine Damage Payments Act 1979 (the “Vaccine
Damage Payments Act” or the “Act”)
Under the Vaccine Damage Payments Act, the UK Government
provides a lump sum payment (currently £120,000) to
individuals severely disabled (defined as at least 60% disability)
as a result of vaccination against certain diseases listed in the
Act. The Act was initially enacted in response to children who had
become severely disabled from the whooping cough (pertussis)
vaccine. The list of diseases to which the Act applies is regularly
updated by statutory instrument, most recently to include COVID-19.
Entitlement to payment under the Act is generally limited to
individuals vaccinated before their 18th birthday or who received a
vaccine at the time of an outbreak of the relevant disease in the
UK. However, payments following certain vaccinations, including
COVID-19, are not limited in this way.3 Unlike the PREP
Act, which applies broadly to vaccines and therapeutics, the
Vaccine Damage Payments Act applies only to certain vaccines.
Claimants who seek compensation in excess of the £120,000
payment under the Act or who wish to claim with respect to a
vaccine that is not listed must file a negligence or strict
liability claim under the Consumer Protection Act 1987
(“CPA,” which implements the EU Product Liability
Directive 85/374/ EEC in the UK). The CPA (in line with the Product
Liability Directive) imposes liability on the producer of a
defective product without needing to establish fault.
Litigation brought against vaccine manufacturers has had limited
success in the UK courts to date. Except in isolated cases where
manufacturing defects have been established, claimants have
experienced difficulty proving both defect4 and
causation.5
People’s Republic of China
The PRC does not have any legislation equivalent to the
PREP Act. Although the Vaccine Administration Law was recently
enacted on December 1, 2019, it does not provide, nor do other
relevant laws and regulations provide, immunity for the
manufacture, testing, development, distribution, administration, or
use of medical countermeasures against pandemics.6
However, both public administrative systems and private tort
liability play important roles in vaccine regulations in the
PRC.
First, the Vaccine Administration Law imposes an
administrative penalty on marketing authorization holders and
manufacturers involved in various Good Manufacturing Practices,
pharmacovigilance, or regulatory violations such as the production
or sale of vaccines that are counterfeit or of inferior quality,
use of illegal production procedures or processes, failure to
established a vaccine electronic traceability system, and failure
to comply with vaccine storage and transportation management
regulations.
Second, the Vaccine Administration Law imposes liability
for death, severe disability, and damage to organs and tissues
caused by an abnormal reaction to vaccination.7 In such
cases, (1) government funds are available and must be used to
compensate the injured for mandatory vaccinations (e.g., Hepatitis
B, poliomyelitis, Baibai Po) and (2) marketing authorization
holders must compensate the injured for voluntary vaccinations
(e.g., flu, HPV). At present, COVID-19 vaccines are classified as
neither mandatory vaccinations nor voluntary vaccinations. Although
no detailed standards or a classification process exist, mandatory
vaccinations are generally proposed by the national health and
finance sectors and then approved by the State
Council.
For damages caused by quality issues of a vaccine, the
marketing authorization holder of such vaccine must bear the
liability.
GOVERNMENT’S ABILITY TO DIRECT
RESOURCES
The US is the only major market with legislation
permitting the government to direct or redirect US resources for
national defense purposes. The EU has recently signaled a desire to
implement similar legislation in response to shortfalls in COVID-19
vaccine production, while the PRC has a variety of regulations,
measures, and precedents it can employ for similar purposes of
product control, allocation, and prioritization.
United States
The Defense Production Act of 19508
(“DPA”)
Congress first passed the DPA in September 1950 in response to
the US’s military unpreparedness for the Korean War. The DPA is
the primary source of Presidential authority to expedite and expand
the supply of materials and services from the US industrial base in
order to support the national defense on a temporary basis. Over
the years, the meaning of “national defense” has been
expanded and now includes, among other things, emergency
preparedness activities conducted pursuant to Title VI of the
Stafford Act; protection or restoration of critical infrastructure;
and efforts to prevent, reduce vulnerability to, minimize damage
from, and recover from acts of terrorism within the US.
The DPA carries two principal authorities. The first is
“rated” or “priority orders,” pursuant to which
the President may compel companies to accept and prioritize
contracts for supplies critical to the national defense. If
received, rated orders must be accepted and performed. Narrow
exceptions apply where performance as ordered is not possible, but
the recipient is generally required to offer the next-best
substitute performance. DPA orders also flow down the
recipient’s supply chain such that subcontractors or suppliers
must prioritize the rated order over competing obligations as well.
Although performing a priority order may require breach of other
contractual obligations, the DPA forecloses civil liability for
damages or penalties resulting from actions taken to comply with
the DPA.
The second authority is “allocation orders,” pursuant
to which the President may compel industry actors to allocate
resources-for example, by reserving manufacturing capability or
supplies in anticipation of a rated order or allocating
manufacturing capability to a particular purpose. This generally
requires proportional allocation across an industry; individual
market participants cannot be singled out.
Failure to comply with a DPA order carries criminal penalty. Due
in part to this legal risk, very few cases are litigated to test
the true scope of authority. Receiving a DPA order may carry
advantages for managing the supply chain and obligations to third
parties, as it provides manufacturers the ability to require their
subcontractors to prioritize the manufacturer’s product over a
competitor’s product.
The Trump Administration used both authorities in response to
COVID-19, primarily by issuing rated orders to compel production of
critical equipment (e.g., ventilators, vaccine and testing
supplies, personal protective equipment (“PPE”)), as well
as using allocation orders to impose very limited restrictions on
exports of PPE and compel ongoing operation of critical factor
operations (e.g., the food supply chain). In many cases, the Trump
Administration preferred to negotiate with the private sector
rather than issue formal DPA orders, but at least some of the BARDA
Operation Warp Speed supply chain contracts were rated orders.
President Biden has issued an executive order authorizing the
heads of the relevant government agencies to use the DPA to fill
shortfalls in COVID-19 response supplies. For more detail on this,
please see our advisory located on our website: https://www.arnoldporter.com/en/perspectives/
publications/2021/01/expanded-use-of-the-dpa.
Although never tested in litigation, the US government has
historically adopted the position that the DPA applies only to US
business operations. This is consistent with a fair reading of the
statute. Accordingly, the DPA likely would not be used to attempt
to compel actions by a non-US corporation acting outside the US,
but may be used to control manufacturing activities taking place
within the US, even if those activities are undertaken by a non-US
corporation.
European Union
The EU does not have any specific legislation equivalent to the
DPA, but individual Member States may have adopted their own
equivalents.
That said, European Council President Charles Michel has
publicly suggested that the EU could use a provision in the EU
Treaties to adopt “urgent measures” in response to a
shortfall in COVID-19 vaccine production. The provision in
question, in Article 122 of the Treaty on the Functioning of the
European Union (“TFEU”), is generally viewed as a
mechanism for the EU to rush emergency financial support for Member
States. According to press reports, the Legal Service of the
Council takes the view that Article 122 of the TFEU could also be
used to force vaccine developers and manufacturers to share
intellectual property or to compel those companies to ramp up
vaccine production for supply to EU Member States.
People’s Republic of China
Similar to the EU, the PRC does not have any legislation
equivalent to the DPA; however, there are regulations,
administrative measures, and judicial precedents in the PRC
empowering the government to control, allocate, and prioritize
production of essential products for epidemic prevention and
control, as described below.
National Plan for Response to Public Health
Emergencies (2006)
The National Plan for Response to Public Health Emergencies
states that governments at all levels must gather all emergency
materials and equipment as necessary in emergency handling. After
the completion of the public health emergency response work, the
government at each level must reasonably evaluate the materials
urgently collected and requisitioned from relevant entities,
enterprises, and individuals and offer compensation.
PRC Vaccine Administration Law
Article 66 of the Vaccine Administration Law recognizes vaccines
to be included in the national strategic material reserves and
requires the state to implement reserves at the central and
provincial levels. Currently, there is no special law or regulation
governing the procurement or restriction on national strategic
material reserves in China.
PRC Drug Administration Law (“Drug
Administration Law”)
Article 92 of the Drug Administration Law provides that the
government is entitled to carry out an urgent drug allocation in
the event of serious disasters, epidemic outbreaks, or other
emergencies- such as COVID-19.
PRC Drug Administration Law (“Drug
Administration Law”)
Article 92 of the Drug Administration Law provides that the
government is entitled to carry out an urgent drug allocation in
the event of serious disasters, epidemic outbreaks, or other
emergencies- such as COVID-19.
PRC Foreign Investment Law
Article 20 of the Foreign Investment Law states that the state
normally does not expropriate the investment of foreign investors.
However, the state may expropriate or requisition the investment of
foreign investors under special circumstances for the need of the
public interest.
Government Notices
On the local level, multiple notices issued by the Chinese
central government in early 2020 require (1) local governments to
take charge of manufacturing epidemic prevention materials and (2)
manufacturers to obey the centralized arrangement. Although they do
not target COVID-19 vaccines or therapeutics, these notices may
indicate an inclination of the Chinese government to control
manufacturing and distribution of such vaccines and
therapeutics.
EMERGENCY USE AUTHORIZATIONS FOR VACCINES AND THERAPEUTICS
Each of the major markets has the authority to temporarily
authorize the distribution of drug products. In the PRC, such
authority is generally limited to vaccines. In the US, EU, and UK,
such authority applies more broadly to medicinal products. The US
government also has the ability to temporarily authorize the use of
approved medicines for unapproved uses.
United States
Emergency Use Authorization
(“EUA”)
The FDA has the authority to permit both approved and unapproved
medical products for unapproved uses to be manufactured and
distributed under specific conditions and labeling during the
period of a declared pandemic or other health
emergency.9 One such condition is that an agent (here,
SARSCoV-2) can cause a serious or life-threatening disease or
condition (here, COVID-19). An emergency declaration invoking the
EUA authority was issued by the Secretary of HHS on February 4,
2020, for COVID-19. Since then, the FDA has issued hundreds of EUAs
for COVID-19-related therapeutics, devices, diagnostics, and
vaccines. If granted, an EUA is in effect only during the period
specified in the declaration and an additional time period
specified in the declaration for ensuring proper disposition of the
product. Thus, an EUA is not a substitute for (and is not intended
to delay) applications for actual clearance or approval. The FDA
can revoke or terminate an EUA at any time.
European Union
Directive 2001/83/EC
Article 5(2) of Directive 2001/83/EC permits the EU Member
States to temporarily authorize the distribution of unauthorized
medicinal products in response to the spread of pathogenic agents,
toxins, chemical agents, or nuclear radiation that may cause harm
to human beings, such as the COVID-19 outbreak. The specific rules
for the implementation of Article 5(2) supply programs are set out
in the national laws of the EU Member States.
United Kingdom
Human Medicines Regulations
Regulation 174 of the Human Medicines Regulations is consistent
with Article 5(2) of Directive 2001/83/EC and permits temporary
authorization of a medicinal product in response to the confirmed
or suspected spread of pathogenic agents. As mentioned earlier, the
Human Medicines Regulations were amended on October 16, 2020, in
response to the COVID-19 pandemic. Among other things, the
amendment strengthened existing provisions that allow for the
temporary licensing of medicines and vaccines. As a result, several
COVID-19 vaccines have been granted temporary authorizations.
People’s Republic of China
Vaccine Administration Law
The PRC’s counterpart to the FDA, the National Medical
Products Administration (“NMPA”), may authorize the
emergency use of vaccines within a certain scope and time period in
the case of any particularly serious public health emergency or any
other emergency that poses a serious threat to public
health-including COVID-19-pursuant to Article 20 of the Vaccine
Administration Law. The application of such emergency use must be
proposed by the National Health Commission as required for the
prevention and control of the infectious disease. However, unlike
the FDA’s emergency authorization powers that apply broadly to
all medical products, the NMPA’s powers are limited to
vaccines.
Under the Vaccine Administration Law and other relevant
regulations, a vaccine that is urgently needed as a countermeasure
against COVID-19 also qualifies for the following expedited
regulatory approval pathways to accelerate its listing and
sales:
- Priority Review and Approval: Both
Article 96 of the Drug Administration Law and Article 19 of the
Vaccine Administration Law permit priority review and approval of
urgently needed new drugs for the prevention and treatment of
serious infectious diseases. - Conditional Approval: Article 20 of
the Vaccine Administration Law permits conditional approval of
vaccines urgently needed in response to a major public health
emergency after being assessed by the NMPA. After receiving a
conditional approval, the vaccine marketing authorization holder is
required to complete the conditional approval criteria for
marketing and post-marketing research work within a specified
period stipulated by the NMPA. - Exemption of Release Approval:
Article 28 of the Vaccine Administration Law provides that, upon an
approval from the NMPA, a vaccine developed in response to an
emergency can be exempted from approval before the release of each
batch to the market.
PRICE GOUGING
The PRC is the only major market with federal legislation that
directly addresses price gouging. The US has typically regulated
price gouging on the state level, while the EU and UK have used
national competition laws to regulate price gouging. However, in
response to the COVID-19 pandemic, each of the major markets has
implemented new guidelines or worked within the parameters of
existing legislation to regulate the pricing of popular pandemic
items like masks and hand sanitizer.
United States
Generally, price gouging in the US has been regulated under
state law. States use a variety of legal authorities and
enforcement postures to target price gouging. The US does not have
a federal price gouging statute. However, on March 25, 2020, the
Department of Justice (“DOJ”) announced that it is using
a provision of Title I of the DPA (referenced above) that prohibits
“hoarding” to target companies that are allegedly price
gouging for PPE, such as N95 masks.10 Apparently, the
DOJ is interpreting the DPA’s hoarding restrictions to also
prohibit charging exorbitant prices for scarce commodities needed
to respond to the COVID-19 pandemic. This is a novel use of the DPA
that has emerged during the pandemic.
For more detail on price gouging, please see our advisory
located on our website: https://www.
arnoldporter.com/en/perspectives/publications/2020/04/covid-19-puts-spotlight-on-price
-gouging.
European Union
Price gouging is addressed by EU authorities using the
competition law rules and, more specifically, the fact that
excessive pricing is an abuse of a dominant position under both EU
competition laws and the national equivalents. During the COVID-19
pandemic, several European countries have regulated the prices of
certain products, such as face masks and hand sanitizer, by setting
maximum retail prices. The European Commission (the
“Commission”) and Member States’ competition
authorities have also indicated that they would closely and
actively monitor the market to detect instances of undertakings [?]
taking advantage of the crisis. As a result, several antitrust
investigations were launched into price increases and output
restrictions of healthcare materials and other products. However,
excessive pricing cases have historically been rare and difficult
for the authorities.
United Kingdom
After the Brexit transition period, UK competition rules are not
expected to change in substance, at least in the short to medium
term. Price gouging therefore continues to fall within the type of
conduct that might constitute an infringement of national
competition rules. In substance, high (or excessive) prices can
give rise to an antitrust violation only if (i) the seller is in a
dominant position, (ii) the price is excessive, and (iii) the price
is unfair either in itself or in comparison with other products.
Since the beginning of the COVID-19 pandemic, the UK competition
regulator, the Competition and Markets Authority (“CMA”),
has made monitoring price increases and tackling any form of price
gouging one of its key objectives. As early as March 2020, an open
letter to the pharmaceutical and food and drinks industries was
issued to warn firms against capitalizing on the current situation
by charging unjustifiably high prices for essential goods.
The CMA has so far opened four investigations into suspected
excessive pricing by a number of UK retailers (including
pharmacies) in relation to the price of hand sanitizer, which had
increased up to almost 400% since the beginning of the pandemic.
Additionally, in June 2020, the CMA issued a joint letter with the
General Pharmaceutical Council encouraging all pharmacies to
“ensure that their prices for essential products,
including hand sanitizer, face masks and paracetamol, do not
include higher than usual mark-ups, when compared to their
pre-coronavirus mark-ups for those products and their mark-ups more
generally.” These investigations into excessive pricing
have now been closed without an infringement finding. Thus, while
the CMA has a high hurdle to overcome to find an infringement, it
is clear that the CMA is actively monitoring the market and is
willing to investigate allegations of excessive prices.
People’s Republic of China
Price gouging is regulated in China through the PRC Price Law
and its implementation rules, as well as competition laws and
regulations. In response to COVID-19, the State Administration for
Market Regulation issued a guideline on the implementation rules of
the PRC Price Law on February 1, 2020. The guideline targets
epidemic prevention products, including masks, antiviral medicine,
disinfection and sterilization products, and relevant medical
devices and equipment, and provides standards and examples for
determining illegal price gouging activities.
COOPERATION BETWEEN COMPANIES
Each major market has implemented a framework to ensure
expedited review of antitrust compliance for companies
collaborating to help combat the COVID-19 pandemic. The US and
European governments have established review procedures to evaluate
COVID-19-related joint ventures and guidance for businesses seeking
to collaborate on health- and safety-related projects during the
pandemic. In the UK, the Competition and Markets Authority has
similarly issued guidance on COVID19-related business cooperation,
while the UK government has provided for a number of exclusions in
specific sectors from the application of UK competition law. In the
PRC, the State Administration of Market Regulation has established
expedited merger reviews, exemptions to certain competitor
collaborations, and heavier penalties on certain antitrust
violations in an effort to facilitate pandemic control and work
resumption.
United States
On March 24, 2020, the US antitrust agencies (FTC Bureau of
Competition and DOJ Antitrust Division) issued a joint
statement11 detailing an expedited antitrust review
procedure and providing guidance for businesses seeking to
collaborate on health- and safety-related projects during the
COVID-19 pandemic.
The agencies announced that they would accelerate their review
of requests for FTC Advisory Opinions and DOJ Business Review
Letters, which are part of a well-established procedure whereby
firms or individuals submit proposals for joint ventures or other
conduct to the agencies for evaluation and receive a response
advising on whether the proposed conduct complies with US antitrust
laws.
The agencies’ responses times can vary, but typically the
procedure takes several months. In the March 2020 joint statement,
the agencies announced that, effective immediately, they would
expedite their review of proposals involving cooperative conduct
related to addressing COVID-19 and its aftermath. Specifically, the
agencies stated that they would “aim to respond
expeditiously” to all COVID-19- related requests for Advisory
Opinions or Business Review approvals, and to resolve those
requests addressing public health and safety within seven calendar
days of receiving all necessary information.
The agencies also signaled that they would “account for
exigent circumstances” when evaluating efforts to
mitigate the spread and impact of COVID-19. The joint statement
recognizes that healthcare providers or facilities may need to
cooperate to provide communities with necessary resources or
services (e.g., PPE, other medical supplies) and that certain
businesses may need to temporarily combine their manufacturing or
logistical capabilities to facilitate the production or
distribution of COVID-19-related supplies: “These sorts of
joint efforts, limited in duration and necessary to assist
patients, consumers, and communities affected by COVID-19 and its
aftermath, may be a necessary response to exigent circumstances
that provide Americans with products or services that might not be
available otherwise.” The agencies also noted that the
antitrust laws already allow for collaboration in many
circumstances (e.g., research and development efforts, sharing
technical know-how and clinical best practices, certain joint
purchasing arrangements) and that “many types of
collaborative activities designed to improve the health and safety
response to the pandemic would be consistent with the antitrust
laws.”
European Union
Temporary Framework for assessing antitrust issues
related to business cooperation in response to situations of
urgency stemming from the current COVID-19 outbreak
(“Temporary Framework”)
On April 8, 2020, the European Commission published a Temporary
Framework for the assessment of horizontal cooperation during the
COVID-19 outbreak. The Temporary Framework sets out the key
criteria the Commission will take into account when assessing
certain types of cooperation and establishes a procedure for the
provision of guidance for specific conduct by way of an ad hoc
comfort letter.
The Temporary Framework applies to conduct that is necessary to
ensure the supply and adequate distribution of essential scarce
products and services, including notably “medicines and
medical equipment that are used to test and treat COVID-19 patients
or are necessary to mitigate and possibly overcome the
outbreak.” The Temporary Framework sets out certain
limited forms of cooperation (e.g., reallocating stocks, switching
production lines, aggregating production, sharing information on
shortage risks) that the Commission does not consider to give rise
to competition issues provided they are subject to sufficient
safeguards.
Under normal circumstances, such measures would in principle be
problematic under the competition rules. However, in light of the
current exceptional circumstances, the Commission does not consider
them to be an enforcement priority, provided the measures are: (i)
objectively necessary to address or avoid a shortage of supply of
essential products of services; (ii) temporary; and (iii) not
exceeding what is strictly necessary to address or avoid the
shortage of supply. The fact that such cooperation is encouraged or
requested by a public authority is also a relevant factor. The
Temporary Framework recommends that companies document their
exchanges and agreements so they can be made available to the
Commission on request.
Additionally, the Commission created a procedure allowing it to
provide ad hoc guidance on specific, temporary cooperation projects
by means of comfort letters. The first comfort letter was issued on
April 8, 2020 to Medicines for Europe, an association of generic
pharmaceutical companies, for a cooperation project involving
information sharing aimed at managing the risk of shortages of
medicines needed in intensive care units for the treatment of
COVID-19 patients. The Commission concluded that the cooperation
was justifiable under the Temporary Framework but imposed a number
of conditions: (i) the project must be open to any interested
pharmaceutical company; (ii) meeting minutes must be kept and
agreements must be shared with the Commission; (iii) only
indispensable information may be shared; (iv) information must be
collected either by the association or a third party; and (v)
information may be shared in aggregate form only.
Informal guidance from the European Commission on specific
initiatives can be found at https://ec.europa.eu/competition/antitrust/coronavirus.html.
United Kingdom
Since the beginning of the COVID-19 pandemic, the CMA has issued
a number of measures responding to the challenges posed by the
pandemic. The key message is that while competition law is very
much still in force, it should not prohibit actions that are
necessary to alleviate urgent situations relating to COVID-19 (such
as permitting necessary coordination in order to ensure the supply
and fair distribution of scarce products to consumers).
In an effort to give businesses greater flexibility to engage in
targeted cross-competitor cooperation, on March 25, 2020, the CMA
issued formal guidance on cooperation during the pandemic. The CMA
reassured businesses that it will not take enforcement actions
against temporary, necessary cooperation aimed at avoiding
shortages or meeting the supply needs of essential products and
services arising as a result of the COVID-19 pandemic. However, it
also stressed that businesses do not have a “free pass”
to engage in anti-competitive conduct and exploit the crisis as a
cover for nonessential collusion. Underlying the CMA’s approach
to enforcement is the potential for coordination to cause harm to
consumers or to the wider economy, particularly where the
collaboration involves coordination on pricing.
In parallel, the UK government has issued, through ad hoc
legislation, a number of exclusions in specific sectors from the
application of UK competition law. In particular:
- In order to assist the UK National
Health Service (“NHS”) in addressing the effects of
COVID-19 on the provision of healthcare services, independent
healthcare providers and NHS bodies have been permitted to, among
other things, exchange information on capacity, share staff and
facilities, and divide activities within particular geographic
areas. - Ferry companies operating services on
the Isle of Wight and the UK mainland have also been permitted to
coordinate on timetables and routes as well as deployment of staff
and vessels. - A package of measures (now withdrawn)
was adopted to alleviate shortages and/or excess demand in the
grocery sector, allowing some cooperation among grocery suppliers
and among logistic service providers-including on staff deployment,
joint purchasing, division of activities in a particular area, and
storage and vehicle capacity. - Another package of measures (also now
withdrawn) temporarily permitted collaboration between dairy
farmers and producers to avoid a surplus of milk-including sharing
information on surpluses, stock, capacity, and demand as well as
coordinating to reduce production.
Importantly, information sharing and/or coordination on prices
and costs have not been permitted under any of the existing or
withdrawn exclusions. It is also worth noting that the above
exclusions are only capable of disapplying UK competition rules; no
such exclusions are available for conduct having an effect outside
the UK, including trade with EU Member States.
To ensure effective monitoring and prompt action with respect to
competition concerns arising from the current crisis, the CMA has
also set up a COVID-19 taskforce as well as a dedicated online
service to report allegedly unfair practices. In this context, the
CMA has been a very valuable point of contact for businesses and
has shown a high degree of pragmatism. In our direct experience of
dealing with the CMA’s taskforce, we have found its dialogue
with companies dynamic and responsive. That said, the key takeaway
is that the CMA is closely watching market developments to monitor
where government action may be required.
People’s Republic of China
On April 4, 2020, China’s antitrust regulator, the State
Administration of Market Regulation (“SAMR”), released a
Notice on Supporting Anti-Monopoly Law Enforcement for Pandemic
Prevention and Control and Resumption of Work and Production
(“Notice”). The Notice, among other things, provides for
expedited merger reviews, exemptions to certain competitor
collaborations, and heavier penalties for certain antitrust
violations.
In its Notice, SAMR established a green channel to expedite
merger reviews in the following areas: (i) sectors closely related
to the control of the pandemic and daily necessities, such as
manufacturers of pharmaceuticals and medical devices, food,
transportation, wholesale, and retail; (ii) sectors severely
impacted by the pandemic, such as restaurants, accommodations, and
tourism; and (iii) transactions that facilitate the resumption of
work and production.
SAMR further indicated that it will exempt certain competitor
collaborations from antitrust scrutiny to help combat the pandemic
and resume work and production, including collaborations to improve
existing technology and to research and develop new products
related to pharmaceuticals, vaccines, test technology, medical
devices, and PPE. SAMR noted that in order to qualify for the
exemptions, companies must continue to meet the requirements under
Article 15 of PRC’s Anti-Monopoly Law, which requires the
companies to demonstrate that the concluded agreement will not
materially restrict competition in the relevant market and will
enable consumers to share the benefits of the collaboration.
SAMR also called for accelerated investigation and heavier
penalties imposed by provincial authorities on antitrust violations
hampering pandemic control and work resumption if the violation
occurs in sectors closely related to the control of the pandemic
(such as manufacturers of masks, pharmaceuticals, medical devices,
and disinfectants), public utilities (such as suppliers of water,
electricity, and gas), and other sectors closely related to
people’s livelihood.
EXPORT CONTROL
Each major market has implemented or updated its export control
regime in response to the COVID-19 pandemic. While each major
market has restricted the export of certain medical supplies, the
EU and PRC have implemented additional restrictions on the export
of COVID-19 vaccines.
United States
The US has the most comprehensive export control regime of the
major markets. Three separate legal regimes currently govern US
exports: (1) the International Traffic in Arms Regulations
(“ITAR”); (2) the Export Administration Regulations
(“EAR”); and (3) new COVID-19-related restrictions on the
export of certain healthcare and medical resources implemented by
the Federal Emergency Management Agency (“FEMA”).
ITAR
Without a license or other authorization, the ITAR restricts the
export, reexport, or in-country transfer of defense articles and
services on the US Munitions List, including hardware, software,
and technical data. Certain biological and biotech-related items
are controlled under the ITAR, but not “biological
agents,” which are certain listed pathogens that have been
non-naturally genetically modified to increase persistence in the
environment or the ability to overcome standard immunity or medical
countermeasures. Sars-Cov-2, whether or not non-naturally
genetically modified, is not currently subject to any controls
under the ITAR. There may be controls on other genetically modified
pathogens used for research.
EAR
Similarly, without a license or authorization, the EAR restricts
the export, reexport, or in-country transfer of commodities,
software, and technology on the Commerce Control List, including
certain biological items. The list also covers chemical agents and
other items that may be used in connection with biotech research.
Unlike the ITAR, the list of controlled biological items includes
naturally occurring pathogens, including many viruses, genetic
elements of such viruses, and vaccines for such viruses. The list
includes “severe acute respiratory syndrome-related
coronavirus (SARS-related coronavirus).”
However, the Commerce Department, which implements the EAR,
considers Sars-Cov-2 to be an entirely distinct biological item.
Therefore, the Commerce Department has issued guidance that
SarsCov-2 is not subject to EAR controls on
“SARS-related coronavirus” and instead is controlled at a
level that would restrict export only to embargoed countries such
as Cuba, Syria, Crimea, and North Korea (a level that applies
equally to essentially any non-listed item in the US, including
pencils, furniture, etc.). Therefore, there are currently no
applicable controls on the export of Sars-Cov-2, its genetic
elements, or related vaccines. The Commerce Department’s
guidance on Sars-Cov-2 indicates that the EAR may eventually
determine that the virus and related items should be controlled at
a higher level where a license may be required for any export,
including to the UK. Furthermore, other biological or chemical
elements of vaccines or therapeutics may be subject to higher
restrictions. For example, certain viruses used to activate genes
in research are subject to higher controls, as are certain viral
vectors used in vaccines. Accordingly, any specific scope of
research should be evaluated before export.
Finally, it is worth nothing that some pandemic-related
equipment can be controlled at a higher level than the products
themselves. For example, certain sophisticated storage tanks that
may be used to transport sensitive pharmacological products may
require a license for export to most destinations because of the
potential use of such tanks for chemical weapons purposes.
COVID-19-Related FEMA Rules Blocking Exports of
PPE
On April 10, 2020, FEMA issued the first of several temporary
final rules allocating “for domestic use” certain PPE
designated as “covered materials” and restricting exports
of “covered materials” without explicit approval by FEMA.
These covered materials include N95 filtering facepiece
respirators; other filtering facepiece respirators (e.g., those
designated as N99, N100, R95, R99, R100, P95, P99, or P100);
elastomeric, air-purifying respirators and appropriate particulate
filters/cartridges; PPE or surgical masks; and PPE or surgical
gloves. The list of items now also includes surgical gowns and
surgical isolation gowns as well as syringes and hypodermic needles
(whether distributed separately or attached together) needed to
administer the COVID-19 vaccines (either piston syringes or
hypodermic single lumen needles with a safety feature). FEMA has
stated that shipments of specified needles and syringes will be
held and returned to the US supply chain only if there is a
critical need for such medical equipment to vaccinate the US
population against COVID-19.
These FEMA export restrictions are subject to a number of
exemptions, including (i) intracompany transfers of covered
materials by US companies from domestic facilities to company-owned
or affiliated foreign facilities; (ii) shipments of covered
materials that are exported solely for assembly in medical kits and
diagnostic testing kits destined for US sale and delivery; and
(iii) sealed, sterile medical kits and diagnostic testing kits
where only a portion of the kit is made up of one or more covered
materials that cannot be easily removed without damaging the kits.
Another exemption allows for exports to customers abroad, which
must meet certain historical supply criteria.
European Union
Regulation (EU) 2015/479 of the European Parliament
and of the Council of 11 March 2015 on common rules for exports
(“Regulation (EU) 2015/479”)
Article 5 of Regulation (EU) 2015/479 governs the common EU
rules for exports and gives the Commission the power to require
authorization for the exports of certain goods. This applies in
critical situations resulting from shortages of essential products.
The Commission used this mechanism during the early part of the
COVID-19 pandemic, subjecting the export of PPE to prior
authorization for the period from March to May 2020. In January
2021, the Commission adopted an export transparency and export
authorization mechanism applicable to COVID-19 vaccines before they
are exported from the EU. The measure is due to end on March 31,
2021.
Commission Implementing Regulation (EU) 2021/111 of
29 January 2021 making the exportation of certain products subject
to the production of an export authorization (“Implementing
Regulation 2021/111”)
On January 29, 2021, the European Commission adopted
Implementing Regulation 2021/111, creating a temporary export
authorization mechanism for COVID-19 vaccines purchased by the EU
under Advance Purchased Agreements (“APAs”).
The Commission has insisted that the new rules are mainly
designed to ensure transparency about the quantities of produced
and delivered vaccines covered by APAs, particularly so that
manufacturers do not export vaccines required to implement the
APAs, without impacting the EU’s international commitments to
equitable access.
The new rules, however, subject exports of vaccines outside the
EU to specific authorizations, which are issued by the competent
authorities of the EU Member States in which the vaccines were
manufactured. Authorizations can be granted only for exports that
do not pose a threat to the execution of the APAs, as concluded by
the Commission on behalf of the Member States.
Authorization requests must be submitted to the individual
Member State for an initial assessment; however, authorization is
overseen by the Commission. Member States must notify the
Commission of applications for export authorizations and provide
draft decisions. If the Commission disagrees with a Member
State’s recommendation, it may issue an opinion, which the
Member State must follow.
Manufacturers of vaccines covered by the APAs must, along with
their first request for authorization, provide a significant amount
of information, including data concerning their exports in the
three months prior to entry into force of the new Regulation (as of
October 29, 2020) (i.e., volume of exports, final destinations and
recipients, and a precise description of the products) as well as
information on the number of vaccine doses under the APAs
distributed in the EU since December 1, 2020, broken down by Member
State.
Some exports are exempted from the new authorization mechanism.
These include: (i) exports of vaccines purchased and/or delivered
through COVAX; (ii) exports to low- and middle-income countries;
(iii) exports in the context of a humanitarian emergency response;
and (iv) exports of vaccines purchased by Member States under the
APAs and resold or donated to third countries.
The new mechanism is expected to remain in force until March 31,
2021, but it can be extended.
United Kingdom
Restrictions on the export of PPE products were implemented by
the UK in March 2020 in accordance with European Commission
Implementing Regulation 2020/568. The Implementing Regulation
expired in May 2020 and was not renewed. Following the end of the
Brexit transition period on December 31, 2020, EU laws no longer
apply to the UK.
The UK government has restricted parallel exports (both to the
European Economic Area and exEuropean Economic Area countries) and
hoarding (i.e., withholding by wholesale dealers) of some medicines
in order to meet the needs of UK patients. The list of products to
which these restrictions apply is regularly updated, most recently
on December 22, 2020: https://www.gov.uk/government/
publications/medicines-that-cannot-be-parallel-exported-from-the-uk.
Wholesalers may continue to withhold medicines as part of stock
management arrangements agreed upon with marketing authorization
holders, which is not considered to be hoarding. They may also
continue to maintain stockpiles built up at the request of the
Department of Health and Social Care in connection with Brexit
preparations.
Import duty and value-added tax payments are waived for public
or charitable organizations (e.g., NHS hospitals) that import
protective equipment, certain medical devices, and other specified
products for use during the COVID-19 pandemic. Goods can be
imported on behalf of a public or charitable organization if they
are to be donated or sold (directly or indirectly) to such
organizations.12
People’s Republic of China
Under the Vaccine Administration Law and the quarantine
regulations, export of vaccines from China requires approval from
the NMPA and quarantine inspection. The State Council can restrict
or prohibit the export of drugs in shortage. Since April 1, 2020,
the Chinese government imposed export controls on
Chinese-manufactured COVID-19 medical devices, including COVID-19
detection reagents, medical masks, medical protective clothing,
ventilators, and infrared thermometers. Exporters must declare that
the products have obtained a medical device registration
certificate issued by the NMPA and meet the quality standard
requirements of the importing country.
Footnotes
1. Health and Human Services Department, Declaration
Under the Public Readiness and Emergency Preparedness Act for
Medical Countermeasures Against COVID-19 (March 17, 2020), https://www.federalregister.gov/documents/2020/03/17/2020-05484/declarationunder-the-public-readiness-and-emergency-preparedness-act-for-medical-countermeasures.
Amendments to the Declaration and HHS Advisory Opinions relating to
the PREP Act can be found at https://www.phe.gov/Preparedness/legal/prepact/Pages/default.aspx.
2. These UK regulations are consistent with EU
legislation, but may diverge from EU law in the future, depending
on post-Brexit changes.
3. GOV.UK, Vaccine Damage Payment, https://www.gov.uk/vaccine-damage-payment/eligibility.
4. For claims brought under the Consumer Protection Act
1987 (“CPA,” which implements the EU Product Liability
Directive 85/374/EEC in the UK) that the product in question was
defective.
5. See, e.g., Loveday v. Renton (No 1) 1990 1 Med
LR”. (holding that the claimant failed to prove on the balance
of probability that pertussis vaccine can cause permanent brain
damage in young children). Causation issues also proved fatal to
the measles, mumps, and rubella group litigation in the late 1990s
and 2000s
6. Unlike the PREP Act, which applies broadly to vaccines
and therapeutics, the Vaccine Administrative Law applies only to
vaccines. Further, the law does not provide immunity for vaccine or
therapeutic liability.
7. Vaccination abnormal reaction, as defined by the
Vaccine Administration Law, refers to adverse drug reactions of
qualified vaccines that cause damage to tissues, organs, and
functions of recipients during a standard vaccination process or
after a standard vaccination, when the parties involved are not at
fault.
8. 50 U.S.C. § 4501 et seq.
9. Federal Food, Drug, and Cosmetic Act § 564, 21
U.S.C. § 360bbb-3.
10. Department of Health and Human Services, Notice of
Designation of Scarce Materials or Threatened Materials Subject to
COVID-19 Hoarding Prevention Measures Under Executive Order 13910
and Section 102 of the Defense Production Act of 1950 (March 25,
2020), https://www.justice.gov/file/1264276/download.
11. Department of Justice, Joint Antitrust Statement
Regarding COVID-19 (May 1, 2020), https://www.justice.gov/atr/joint-antitrust-statementregarding-covid-19.
12. GOV.UK, Guidance: Pay no import duty and VAT on
medical supplies, equipment and protective garments (COVID-19)
(March 31, 2020), https://www.gov.uk/guidance/pay-no-import-duty-and-vat-on-medical-supplies-equipment-and-protective-garments-covid-19.
The content of this article is intended to provide a general
guide to the subject matter. Specialist advice should be sought
about your specific circumstances.